Quality control of assembled contigs
Go to the relevant folder in the local directory where the assembled bidirectional contigs are stored. Select and open an assembly.
Manually editing assemblies
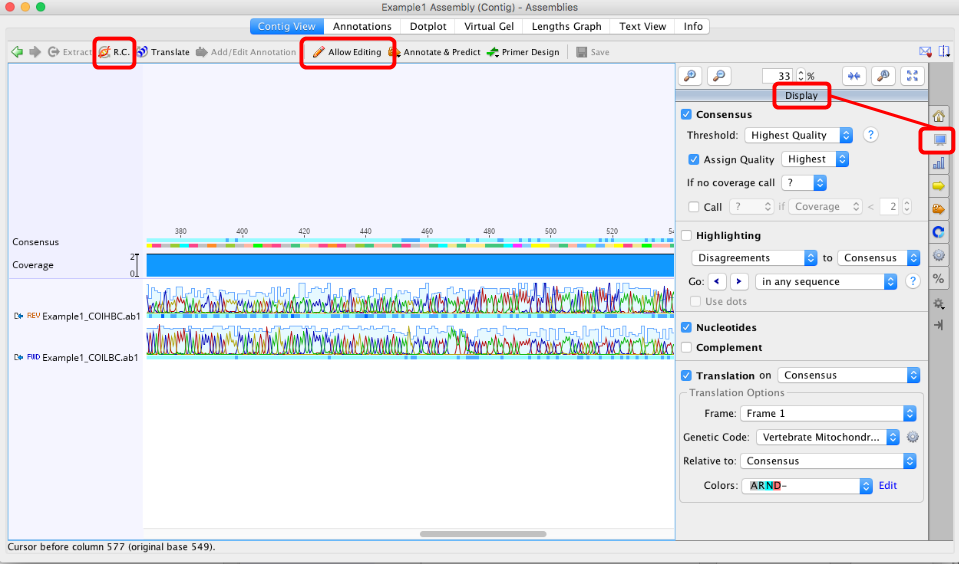
If you are editing protein coding genes, check the “Translation” option in the right hand menu of the Display window
Set the correct genetic code (“Vertebrate Mitochondrial” or “Invertebrate Mitochondrial” for COI, or “Bacterial” for rbcL and matK) and
Select the correct reading frame. Black dots = stop codons, so we do not want any of these. If stop codons are present double-check the following:
the correct genetic code is selected,
the assembly is in the correct orientation (Use “R.C.” button in top left of contig window if you need to reverse complement it),
whether insertions or deletions are present, and/or
check BLAST to verify it is not a contaminant
Quickly scan through the individual assemblies and assess whether or not each disagreement (if present) needs a manual edit.
A manual edit ONLY needs to made if you feel the consensus sequences has been called incorrectly (or there is a gap that needs to be deleted). If Geneious calls the consensus sequence correctly, NO changes should be made to individual traces.
To manually edit an assembly, the “Allow Editing” button in the toolbar of the contig window should be clicked on (see image above).
If you are unhappy with the trimmed portions, you can edit these manually by clicking on, and dragging, the red bar indicating the trimmed region.
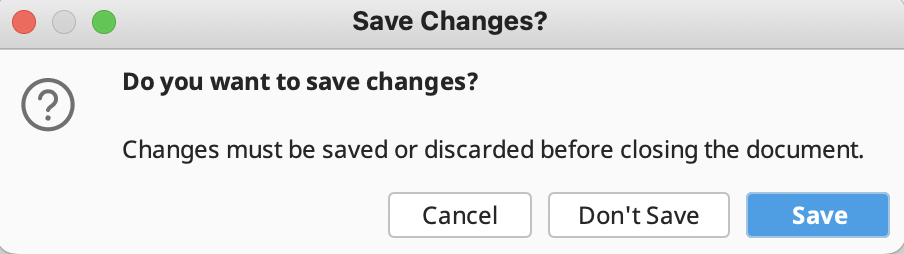
Do not forget to save your edits. You will be prompted to do this when you try to close the assembly.
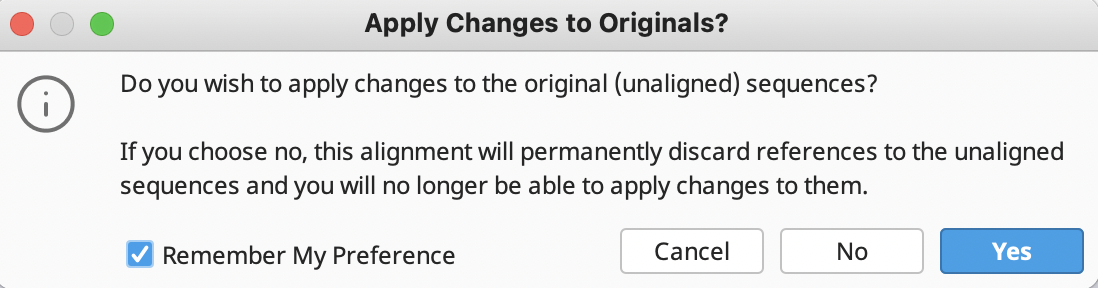
In addition, another prompt window will ask if you want to apply changes to the original sequences. ALWAYS Click “Yes”, because you risk losing connection to reference sequences.
Checking sequence quality with alignments
A second quality check is made by aligning your sequences based on the gene - align COI sequences together, rbcL together, etc.
For alignments of protein coding regions Geneious’s Translation Alignment program doesn’t work like it should, so use an alternate program such an online program called TranslatorX (http://translatorx.co.uk) to create an alignment.
Note
It’s important to note that TranslatorX only checks the forward reading frames, so you need to Reverse-Complement the matK sequences before putting them into this alignment program, otherwise you will receive errors.
Export the consensus sequences (of good assemblies only) as a FASTA file then import this file into the program. We suggest you leave the Protein Alignment Option method selected as “Muscle”. In the Genetic Code box select the relevant reading frame and be sure to check the “Guess most likely reading frame” option. Then hit Submit Query.
If the program runs OK and doesn’t encounter any errors, it will return an alignment of the nucleotides and also an alignment of the amino acids. You may download the fasta file of both, however, the alignment of amino acids is what will be used for the second quality check. Import the fasta file(s) of the alignments into Geneious for further analyses.
Use the alignment to address any issue that you can see i.e. a clear difference between one sequence to the others (Remember this can be possible if the sequences are distantly related but still cross reference the alignment to the individual assemblies). Also, gaps must be assessed and resolved. Major differences in the alignment may also indicate that one or more of the sequences are contaminants (use BLAST to determine this).
You may need to repeat the alignment step a number of times as you cross reference the assemblies and make edits. Save the edits, re-export all the consensus sequences and create a new alignment with these new consensus fasta files.
If more than a handful of edits need to be made to the consensus sequence, the assembly should be discarded and the sample re-sequenced. You need to make a judgement call on this.
BLAST
Geneious provides the ability to BLAST your sequences from within the program. You can read more about BLAST on the NCBI BLAST website here: http://blast.ncbi.nlm.nih.gov/Blast.cgi.
To use BLAST, follow these directions:
Select a single or batch of contigs you want to compare to the NCBI public DNA sequence database – we recommend small batches of less than 15 sequences, since the process can be quite slow otherwise – and click on the “BLAST” button in the Toolbar.
The “BLAST” window appears and has multiple options for consensus, GenBank database, program etc. The selections in the image below are our recommendations for querying COI sequences, however we encourage all users to look into the various options and decide what works best for your dataset.
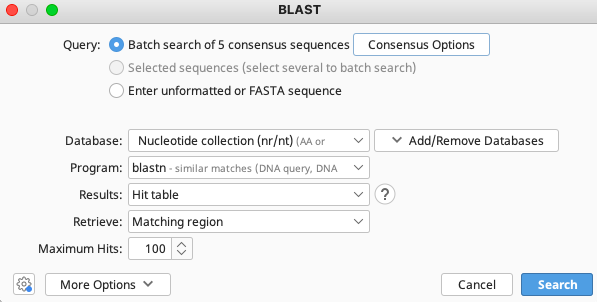
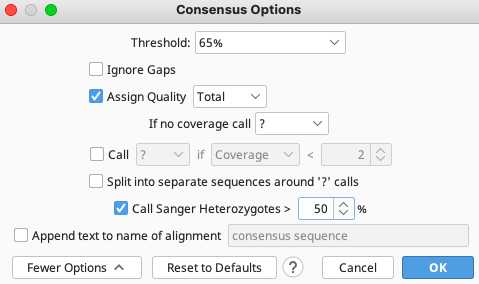
The consensus options allows you to choose how the program will call the consensus sequence of each assembly.
Once you have made your selections, click “Search” button in the “BLAST” window.
The search progress appears in the Document Window. If this is too slow, or you want to exit the search for whatever reason, click on the “Stop” button in the top left of the Document Window.
Once complete, the results are saved in a subfolder (folder name ends with “- nr Megablast”) within the folder containing your query sequence(s). If you did a batch search, there will be further subfolders containing BLAST results for each of the sequences you entered into the BLAST search.
In the results folder the BLAST results are displayed in the “Hit Table” tab. Various information is included e.g. Hit Accession number, Query coverage, % Pairwise Identity, etc. You can choose what is displayed by clicking on the manage columns icon found in the upper right of the table. Further information is found in the other tabs of the folder (Query Centric View, Annotations, Distances, Info).
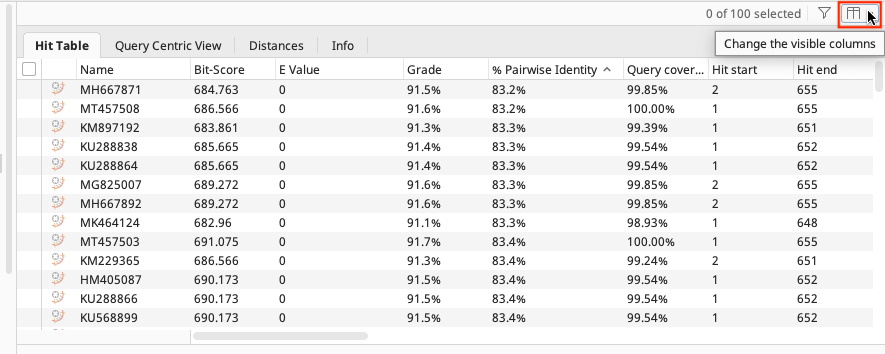
To get more information about the individual BLAST hits, select one of the hits and the information about that sequence appears in the Document Viewer. Any of the columns can be sorted, rearranged, or resized as usual.